Teilen Sie dies:



Rokitnasky-Küster-Hauser-Syndrom Symptome, Ursachen und Behandlungen
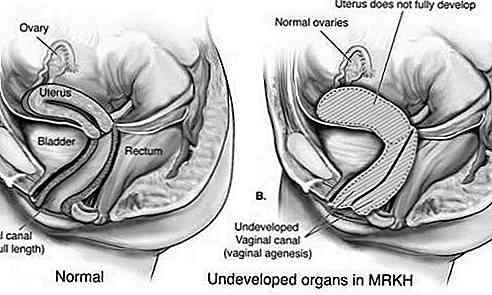
Rokitansky-Küster-Hauser-Syndrom (MRKH) ist eine Störung, die das weibliche Fortpflanzungssystem betrifft, das durch Unterentwicklung oder das Fehlen von Gebärmutter und Vagina gekennzeichnet ist.
Frauen mit diesem Syndrom entwickeln sekundäre Geschlechtsmerkmale während der Pubertät -senos, Schamhaare- haben jedoch keinen Menstruationszyklus (primäre Amenorrhoe).
Der Beginn des Menstruationszyklus ist oft das erste Anzeichen eines MRKH-Syndroms. Obwohl Frauen mit dieser Erkrankung keine Schwangerschaften haben können, wenn sie durch assistierte Reproduktion Kinder bekommen können.
Die Schwere des Syndroms kann abhängig vom Typ variieren. Typ 1 ist durch eine isolierte Abwesenheit der proximalen zwei Drittel der Vagina gekennzeichnet. Typ II wird durch andere Defekte wie Nierenanomalien (40% der Fälle), Skelettanomalien (20-25%), Hörstörungen (10%) und seltene Herzfehler aus.
Aufgrund der Art der Erkrankung ist es wahrscheinlich, dass es zu erheblichen psychologischen Problemen führt, daher wird empfohlen, um Hilfe zu bitten.
Das MRKH-Syndrom hat laut verschiedenen Studien eine weltweite Inzidenz von 1 von 4500 Geburten von Frauen.
Es wird in der Regel häufiger in der Jugend diagnostiziert, wenn es überprüft wird, ob der Menstruationszyklus sich entwickelt. Obwohl Frauen keine Kinder haben können, sind ihre Eierstöcke normal und funktionell.
Symptome des Rokitnasky-Küster-Hauser-Syndroms
Die Symptome des MRKH-Syndroms sind von Frau zu Frau sehr unterschiedlich. Denken Sie also daran, dass betroffene Frauen möglicherweise nicht alle der unten genannten Symptome haben.
Mayer-Rokitansky-Hauser-Typ-I-Syndrom
Dieser Typ ist auch als Müller-Aplasie bekannt und zeichnet sich durch eine unzureichende Entwicklung von Uterus und Vagina aus. In den meisten Fällen haben sich der Uterus und / oder die Vagina nicht entwickelt (Aplasie); oder es besteht eine Verengung des oberen Teils der Vagina und des Uterus (Atresie). Die Eileiter können ebenfalls betroffen sein.
Einige der Merkmale oder Symptome des MRKH-Syndroms sind:
- Erste Menopause oder fehlende Perioden während der Pubertät. Obwohl dieses Symptom beim MRKH-Syndrom häufig vorkommt, erfährt die Patientin eine Pubertät mit normalem Telarca und Adrenarche. Es gibt jedoch keine Menstruation.
Da die Funktion der Eierstöcke normal ist, erfahren Patienten alle Veränderungen, die mit Menstruation oder Pubertät verbunden sind.
-Normale äußere Geschlechtsorgane.
- Reduzierte Vaginaltiefe von 2 bis 7 cm.
- Sexuelle Merkmale wie normale Brüste und Schamhaare.
- Eierstöcke funktional mit normalen Östrogenspiegeln.
-Normale chromosomale Modelle.
-In MRKH Typ 1-Syndrom, nur die Vagina und Gebärmutter sind abnormal. Bei Typ II MRKH kann auch Defekte in der Vagina und Uterus durch Anomalien in dem Eileiter begleitet werden, wie es in den Nieren oder die Wirbelsäule.
- Obwohl Herzanomalien selten sind, können das Aorten-Lungenfenster, der Vorhofseptumdefekt und die Pulmonalklappenstenose auftreten
-Patienten mit MRKH-Syndrom klagen in der Regel über zyklische abdominale Schmerzen aufgrund einer Ablösung des zyklischen Endometriums ohne einen patentierten Entwässerungsweg.
-Esterilität: Viele Patienten suchen oft klinische Behandlung für Unfruchtbarkeit, aber nicht für primäre Amenorrhoe.
- Ein anderes Symptom ist die Schwierigkeit in den sexuellen Beziehungen, da der Grad der vaginalen Aplasie von völliger Abwesenheit bis zu geringer Entwicklung variieren kann, was Dyspareunie verursachen kann.
-Schwierigkeiten des Urinierens, Harninkontinenz, wiederkehrende Harnwegsinfektionen.
- Wirbelanomalien.
- Auf der Ebene der Peritonealreflexion kann eine tastbare Gewebeschlinge vorhanden sein.
- Nierenmalformationen.
Symptome des MRKH-Syndroms Typ II
Nierenversagen ist die häufigste Anomalie im Zusammenhang mit MRKH Typ II-Syndrom.
Frauen mit MRKH-Syndrom Typ II können keine Niere, Fehlbildungen der einer oder beiden Nieren (renale Dysplasie), die Entwicklung von Nieren (Hypoplasie) und / oder falsche Platzierung im Körper eines oder beiden Nieren (Nieren ectopia).
Diese Anomalien können Nierenwachstumsmangel verursachen, Nierensteine, erhöhte Anfälligkeit für Infektionen der Harnwege und abnormale Ansammlung von Urin in der Niere durch Obstruktion.
Viele Frauen mit Typ-II-MRKH Syndrom können auch Skelettfehlbildungen wie Knochenprobleme in dem Hals- und Brustwirbel, die nicht ordnungsgemäß entwickeln können.
Abnormalitäten des Gesichts können auch auftreten, wie beispielsweise eine abnormal kleine Backe (Mikrognathie), Lippen- und Gaumenspalte, und Unterentwicklung von einer Seite der Fläche, in Gesichtsasymmetrie führt.
Viele betroffene Frauen können auch Hörprobleme entwickeln, hauptsächlich aufgrund von strukturellen Anomalien des Mittelohrs.
Wenn die Ohren beteiligt sind, kann die Erkrankung Genital-Nieren-Ohr-Syndrom (GRES) genannt werden.
Einige der Frauen mit MRKH-Typ-II-Syndrom hatten zusätzliche körperliche Anomalien, einschließlich Defekte in den Händen und / oder Armen.
Diese Abnormalitäten in den Extremitäten können das Fehlen eines oder mehrerer Finger und Zehen, einen doppelten Daumen und das Fehlen des langen und dünnen Unterarmknochens (fehlender Radius) umfassen. Nicht alle Symptome treten bei allen Patienten auf, dies hängt vom Ort und der Schwere ab.
Ursachen
In den meisten Fällen ist der Ursprung des MRKH-Syndroms unbekannt und tritt bei Frauen ohne Familienanamnese auf.
Die Forscher weisen auf genetische und umweltbedingte Faktoren als Ursache des Syndroms hin, obwohl noch kein Gen oder Gene, die mit der Krankheit in Verbindung stehen, bestimmt wurden.
Historisch gesehen, haben Forscher vorgeschlagen, dass das Syndrom als Folge von Krankheit oder mütterlichen fetalen Exposition gegenüber verschiedenen schädlichen Substanzen, wie bestimmte Medikamente auftreten können, Missbrauch Drogen, Alkohol ...
Es gibt jedoch keine Studie, die diese Assoziation zwischen dem Syndrom und dem Konsum einiger anderer Medikamente bestätigt.
In einigen Familien scheint das Syndrom ein dominantes Vererbungsmuster zu haben. Das Vererbungsmodell ist jedoch schwer festzustellen, da nicht alle Frauen, die daran leiden, die gleichen Symptome haben, auch wenn sie aus derselben Familie stammen.
Den Forschern zufolge handelt es sich höchstwahrscheinlich um eine Kombination genetischer und umweltbedingter Faktoren.
Was die Anomalien im Reproduktionsprozess betrifft, so sind sie auf eine unvollständige Entwicklung des Müllerschen Ganges zurückzuführen, aber seine Ursache bleibt unbekannt.
Es scheint, dass in den letzten Jahren die Evidenz zugenommen hat, da das MKRH-Syndrom eine genetische Störung ist. Die Zunahme der Fallstudien hat dazu geführt, dass diese Idee weiter gestärkt wird.
Wir sprechen über genetische Störungen, wenn es eine Kombination von Genen für ein bestimmtes Merkmal gibt. Falls es sich um eine dominante genetische Störung handelt, wie es in diesem Fall der Fall wäre, würde sie durch die abnormale Entwicklung einer einzelnen Kopie eines abnormalen Gens erzeugt werden.
Dieses defekte Gen kann sowohl von der Mutter als auch vom Vater vererbt werden, oder es kann auf eine neue Mutation im Gen selbst zurückzuführen sein.
Die Wahrscheinlichkeit, dass dieses defekte Gen übertragen wird, beträgt 50% für jede Schwangerschaft, unabhängig vom Geschlecht des Kindes.
Polygene Vererbung wurde ebenfalls als Ursache für das MRKH-Syndrom vorgeschlagen.
Bis heute wurden bei mehreren vom MRKH-Syndrom betroffenen Personen sieben Deletionen und eine Verdoppelung von Chromosomensegmenten gefunden. Aber nur eine dieser Anomalien wurde pro Person gefunden.
Gegenwärtig sind diese Chromosomen identifiziert worden, wo es möglich ist, dass Segmenteliminierungen beteiligt sind. Diese Chromosomen sind: Chromosom 1 (1q21.1), 4 (4q34q), 8 (8p23, 1), 10 (10p14-15), 16 (16p11.2), 17 (17q12) und 22 (22q11.21) und die Verdopplung wurde auf dem X-Chromosom gefunden (Xpter-p22.32).
All diese neuen Informationen hat Forscher führten mehrere Kandidatengene zu wählen, einschließlich: HNF1B, Lhx1, Tbx6, ITIH5 und SHOX.
Behandlung
Das Ziel der Behandlung ist, dass der Patient eine vollständige und befriedigende sexuelle Funktion hat.
Da die Symptome so unterschiedlich sind, ist die Zusammenarbeit eines Teams von Spezialisten erforderlich, um einen umfassenden Behandlungsansatz sicherzustellen.
Im Allgemeinen sind sie diejenigen Patienten mit dem Syndrom MRKH ermutigen psychologische Hilfe nach der Diagnose zu suchen und vor der Behandlung, da dieses Syndrom Angst oder psychische Belastung verursachen kann. Auch Support-Gruppen werden empfohlen.
In Bezug auf die Behandlung der vaginalen Aplasie ist es, eine neue Vagina zu schaffen.
Die Behandlung kann chirurgisch sein oder nicht. Die nicht-chirurgische wird immer die erste Option sein. Eine der Optionen sind Vaginaldilatatoren, die dazu dienen, zu vergrößern oder eine Vagina zu schaffen.
Die bekannteste Methode ist der Franck-Dilatator, der auch als Dammstrecker bezeichnet wird und keine chirurgische Operation erfordert.
Selbstverwaltung der Patient muss ausreichend motiviert sein, um es zu benutzen. Es dauert ungefähr sechs Wochen bis einige Monate.
Eine andere Möglichkeit kann eine Vaginoplastik sein, die eine flache Vagina erzeugen würde. Aber über diese Operation gibt es nicht viel Übereinstimmung darüber, welche Techniken zu verwenden sind.
Die McLndoe-Technik ist eine der am häufigsten verwendeten chirurgischen Verfahren zur Rekonstruktion der Vagina. Ein Hauttransplantat, das aus dem Gesäß oder dem Oberschenkel entnommen wird, wird entnommen und auf eine penile Prothese aufgebracht, die aufblasbar ist. Diese Transplantatprothese mit den Formen zu vaginal Tunnels, der in schneidend und stumpfen mit einem Skalpell und sezierte öffnet, mit Sorgfalt nicht vegija, Rektum oder Peritoneum zu beschädigen.
Es ist sieben bis zehn Tage und wenn es geändert wird, ist es unter Anästhesie. Später wird der Patient andere Dilatationsprothesen verwenden. Nach drei Monaten kann der Patient Sex haben.
Das Problem bei dieser Technik ist, dass zusätzlich zu Narben eine Langzeitdilatation erforderlich ist.
Eine andere Technik, die für das MRKH-Syndrom verwendet wird, ist die intestinale Neovagina.
Diese Technik verwendet ein isoliertes Segment des Darms. Es besteht darin, ein Fragment des Sigmas durch einen Bauchschnitt zu schneiden und es in den Bereich zu übertragen, in dem der Raum der Neovaginitis geschaffen wurde. Diese Technik ist sehr komplex und kann bis zu 8 Stunden dauern.
Der Vorteil dieser Technik ist, dass sie eine geräumige Vagina von ausreichender Länge hinterlässt und dass sie selbstschmierend ist. In Bezug auf die Nachteile wäre das erste Ding, dass es eine komplexe und infrabominale Chirurgie ist, es lässt überschüssige Schleimsekrete durch das Darmfragment, Schleim vaginalen Prolaps und Entzündung der Schleimhaut in der Neovagina.
Schließlich ist eine andere Technik die Vecchietti-Technik.
Diese Technik übt einen kontinuierlichen progressiven Druck durch eine Acrylolive durch die neovaginale Raumenergie und die Bauchwand aus. Dann wird ein Traktionsgerät in die Peritonealhöhle platziert und entfernt allmählich das Scheidengewölbe. Diese Technik wird jetzt durch Laparoskopie durchgeführt.
Referenzen
- https://visualsonline.cancer.gov/
- https://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrom
- http://emedicine.medscape.com/article/953492-overview
- http://www.news-medical.net/health/Mayer-Rokitansky-Kuster-Hauser-(MRKH)-Syndrom-Symptoms-(Spanish).aspx
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3109
- https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome/
- http://www.biomedcentral.com/content/pdf/1750-1172-2-13.pdf
- Quellbild: http://www.mrkhnorge.org/mrkh/?lang=en